Important Intramolecular HOMO/LUMO
Mixing
(Preview of "Conjugation" and "Resonance"
Stabilization)
We know that mixing an unusually high HOMO with an unusually
low
LUMO can lead to bond formation between two molecules. A similar kind
of interaction can make certain molecules more stable than a naive
person might think they should be. This is the so-called "resonance"
stabilization.
|
Remember our earlier discussion of the special stability
of amides, a stability that we associated with our ability to draw
these two more-or-less reasonable bonding pictures for this functional
group. The picture on the left with complete octets and no separation
of charge is expected to be a more important "resonance structure" than
the one on the right, which also has complete octets but suffers from
charge separation.
[Here we have included two curved arrows for bookkeeping
purposes to show the source of the + charge on N, the C=N double bond,
and the negative charge on oxygen in the minor resonance structure.]
|
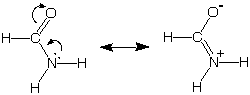
|
Why should our inability to draw a good single
"Kekulé"
picture of this molecule have anything to do with its
stability?
Here again is a case of "Compared to what?"
When we say that an amide is "resonance stabilized," we mean
that
it is more stable than we would have expected if we thought its
properties would be properly predicted on the basis of the first
structure alone, so that the electronic energy would be the sum of
the energies expected for an unshared electron pair orbital on N, a
σ and a π
bonding orbital between C and O, 2 σ
N-H
bonding orbitals, etc.
"Resonance energy" is rather like "correlation energy." Both
are
fancy names for errors we made when we chose too
simple a
model for reality. Both are cases where, by doing what comes
naturally, electrons outsmart us and find a lower-energy home than we
had built for them. [One might imagine an analogy to a public
redevelopment authority that constructs a utopian housing development
for individuals who turn out to be able to do a better job devising
their own homes.]
When we draw the structure on the left and naively suppose
that it
is an adequate structure, we suppose that the energy of the molecule
includes the energy of an unshared pair of electrons on N. Notice
however that the C atom adjacent to this N is part of an unusually
low LUMO, the π* orbital
of the C=O double
bond. The p orbital of the C atom is a larger
contributor to
the antibonding π*
orbital than the
p orbital of the O atom (which was mostly used up in
the
bonding π orbital), so
there is good
overlap between the π*C=O
orbital and the N
lone pair.
In this simple picture there is an unusually low "LUMO" in
position to overlap effectively with the high "HOMO" of the N atom.
In truth the electron pair should not be unshared, as suggested by
the single picture. It should be shared with the adjacent C=O group
and be stabilized in the process. This is why the true molecule is
more stable than we would have guessed on the basis of a single
simple structural diagram.
Notice that this does not involve any crazy bouncing back and
forth between two different positions for the electrons. It is just
that the true molecular orbitals involve a more extensive sharing and
stabilization than the simple picture envisions. It is crucial that
there be effective overlap between the AOs of the carbon and nitrogen
atoms to allow this special stabilization.
The magnitude of the resonance stabilization in the amide
group is
about 16 kcal/mole. This is about 1/4 as large as the energy of the
C-N σ bond itself, so it
is quite
important. This phenomenon is particularly significant in
proteins where every amino acid unit is linked to
the next by
an amide group.
|
Think of some of the other implications of this phenomenon for the properties of proteins.
|
|
GEOMETRY
: Whereas most molecules have easy rotation about single bonds, rotation
about the C-N bond in amides is difficult, "because" there is
a partial double bond, as shown in the ionic resonance structure. The
barrier to this rotation is ~16 kcal/mole (determined by nmr
spectroscopy, as we'll discuss later).
Of course the real problem is that twisting about the
C-N bond destroys the overlap between the unshared pair of the N and
the π* vacant orbital on
C, as shown in the diagram on the right. This turns off the resonance
stabilization and allows us to equate the barrier to rotation with the
magnitude of resonance stabilization.
A second geometric implication has to do with the bonds
to N. We know that the N in NH3 is pyramidal, so
that the N can use some of its s-orbital character
to accommodate the unshared pair. In the amide group, this pair of N
electrons spends some of its time on C=O, so there is no longer as much
reason for the N to use its s-character to
stabilize this pair. A planar arrangement around N
allows the s character to be taken from the pair
and used to form stronger σ
bonds to carbon and the two hydrogens. Furthermore, the N unshared pair
overlaps better with C=O when it is in a p orbital
than when it is in an sp~3
hybrid.
Because there is no "twist" about the central C-N bond,
and the N is sp2 hybridized, all of
the six atoms in the core of the amide group (N-C=O and the
three other atoms attached to C and N) lie in a common plane.
This imposed rigidity has profound implications for the shape of
proteins.
There are also implications for bond lengths.
To the extent that nN (nitrogen's lone
pair) mixes with π*C=O,
the C=O bond should be weakened and lengthened, while the C-N bond
should be strengthened and shortened. These changes are suggested by
the ionic resonance structure for the amide. The average C=O
bond length in 481 aldehydes and ketones is 1.20Ĺ,
while that in 410 amides is 1.23Ĺ, 0.03Ĺ
longer.
The average C-N
bond length in 115 amides is 1.33Ĺ, much shorter than that
the 1.47Ĺ average of 1200 amines, but here there is a
"compared to what" problem, because there is another source of
shortening and strengthening for the amide - the carbon and the
nitrogen atoms both use sp2 rather than sp3
hybrids in making the C-N σ bond.
|
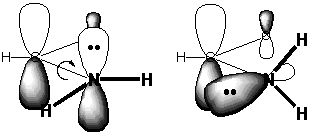
Good Overlap - - - - - - - - -> Poor Overlap

|
|
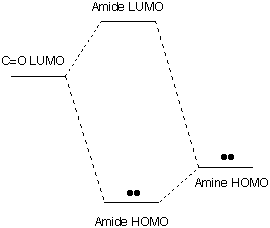
REACTIVITY
: The unshared pair on N, which would have been an unusually high HOMO
in a normal amine, is lowered in energy, and made less reactive, by
mixing with the π* LUMO
of the C=O group.
At the same time the unusually low π* LUMO of the C=O group, which
made it acidic, is raised in energy by mixing with the occupied p
orbital of N. Thus both the basic amine group and the acidic
carbonyl group are made less reactive by interacting within
the amide group.
This is why it is much better to regard the amide group
as a distinct functional group, than as an amine
and a carbonyl group.
The reduction of reactivity makes proteins resistant to
reaction. So when you take a shower, or get a little sodium hydroxide
solution on your hands, you don't have to worry about your skin
dissolving.
|
|
|
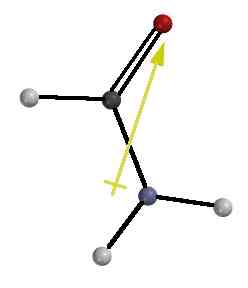
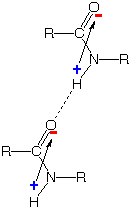
CHARGE
DISTRIBUTION : Shift of the electron pair from N
to C=O gives the amide group a polar character.
This polarity is indicated by the gold arrow in the figure to the left (calculated by MacSpartan Plus). The
+ end of the arrow shows the positive end of the electrical dipole near
N and the point shows the negative end near O.
[The dipole moment
measured experimentally by Kurland and Wilson in 1957 is 3.71 D and is
oriented at 39.6° from the C-N axis, in good agreement with the
calculation. This strength of dipole moment would result from shifting
1/3 of an electron's charge from nitrogen to oxygen.]
Positive-negative attraction between the
charges generated by this electron shift strengthens "hydrogen bonds"
which hold the N-H portion of one amide group to the O atom of another,
as shown on the far right. This kind of association is crucial to the
formation of α-helices, β-sheets, and other "secondary"
structures of proteins.
|
|
Usually we think about HOMO/LUMO mixing being
intermolecular, and leading to reaction between two
different
molecules. Here we think of intramolecular
"HOMO/LUMO" mixing
as a correction to an oversimplified view of the energy, structure,
reactivity, and electron distribution of a single molecule.
Fortunately we can ignore intramolecular mixing for normal bonds and
antibonds because overlap is small and energy-match is poor.
copyright 2001-2003 J. M. McBride