The LUMO of
CH3OH2+
Question 11a of the first semester final exam for
Chem 125a for 2001-2002 was:
|
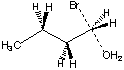
Reaction of 1-butanol with HBr involved
protonating the OH group to give the molecule a lower LUMO,
and then attacking it with the HOMO of Br- to
give a transition state like the one on the right, where the
colinear dotted lines show the forming Br-C bond and the
breaking C-OH2 bond.
|
|
|
a) Explain why protonation lowers the
energy of the LUMO, and draw a picture of the LUMO to
explain the direction from which it is attacked by
Br-, and why the dotted lines should be roughly
colinear.
|
The expected answer for the question
was:
|
Protonation generates a positive charge
(on the Oxygen atom), which lowers the electron energy for
all MOs in the vicinity, including that of the LUMO.
The LUMO is
s*C-O (see right). Best
overlap is available from the face of the carbon atom
exactly opposite the O, as shown by the arrow.
|
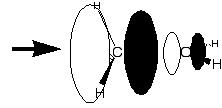
|
This answer was given full credit, but it
does not contain the whole truth.
We can often gain a very good understanding of
molecular structure and reactivity by considering simple localized
orbitals, like s*C-O
shown above, that involve only two atoms. Gaining this kind of
insight was a major goal of Chem 125a, and it should prove very
useful in understanding the reactions you encounter in Chem 125b.
But you should remember that such simple orbitals
are often "disguised" by mixing with other orbitals of similar energy
to give complex MOs that include contributions from many atoms. In
such instances the "true" LUMO (or HOMO) that is calculated by a
quantum mechanics program, like MacSpartan, shows us more than is
convenient to see for the purpose of achieving a simple
understanding. ("I
wanted to catch a little
one!")
The LUMO of
CH3OH2+
is a good example. The figure below shows three different structures
of this molecule and, below each, the corresponding molecular LUMO.
Structures and LUMOs were determined
using STO 6-31G**, the most sophisticated calculation available
with MacSpartan. In each structure the methyl group's third H is
obscured by the black C atom. A single contour (where the electron
density would be 0.001 e/Å3) is shown for each
orbital. Red and blue code opposite signs. Unfortunately
MacSpartan exchanged red and blue for the orbital shown in the
center. For comparison with the flanking MOs, you must pretend
that its colors are swapped.
Images in the center are for the cation in its
lowest-energy geometry. The flanking structures are distorted, as the
molecule would become when it approaches the transition state for
reaction.
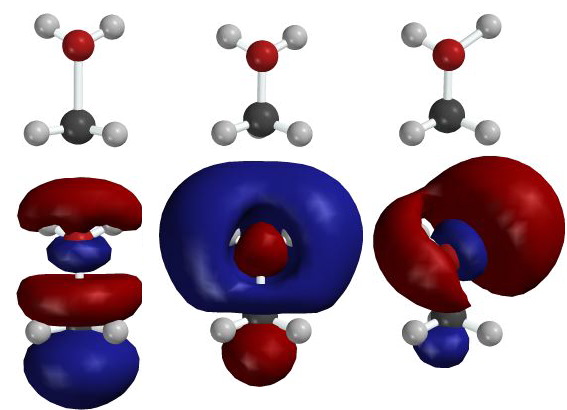
The structure on the left is the
lowest-energy structure possible if the central C-O bond is
artificially stretched by 30%, as it would be if Br-
were approaching from the bottom, mixing its HOMO with
s*C-O,
and causing the C-O bond to break. (Note that the CH3
group has become more planar than in the central
structure.)
The structure on the right is the
lowest-energy structure possible if the O-H bond on the right is
artificially stretched by 30%, as it would be if Br-
were approaching from the top right, mixing its HOMO with
s*C-H,
and causing the C-H bond to break.
The flanking LUMOs are relatively simple. Each is
dominated by the s*
orbital between two atoms whose bond is being broken for that
particular structure, with just traces of the other two localized
s*
orbitals.
The center LUMO, for undistorted
CH3OH2+,
is formed by mixing these two LUMOs with the right-to-left mirror
image of the LUMO on the right, that is, the
s*
orbital whose occupancy by electrons would weaken the other O-H
bond.
Apparently the three localized
s*
orbitals, involving the individual pairs of atoms, O-C, O-H(1), and
H(2)-O, are similar enough in energy that the small overlap among
them can cause this confusing mixture.
Not to worry however.
When the structure approaches the transition state
(and for understanding rate, the transition state energy is what
we need to predict!) the s*
orbital of the breaking bond is much lower in energy than the other
two (because of reduced overlap), and three localized
s*
orbitals no longer mix strongly with one another. So a single
localized pairwise orbital dominates the LUMO of the important
structure.
Thus the simple-minded pair-wise orbital
approach we took really does work for understanding this
reaction.
The shape of the LUMO for undistorted
CH3OH2+
reminds us that three paths are possible for reaction with
bromide. The HOMO of Br- can attack either at an H to form
H-Br and "displace"
CH3OH, or it
can attack at C to form CH3Br and displace
H2O. In
fact, as the shape of the LUMO might suggest, the former reaction,
deprotonation, occurs much more easily, but it is a "nothing"
reaction, since HBr can just protonate
CH3OH again
and again, waiting for the opportunity to displace
H2O and form
the ultimate products.
copyright 2002 J. M. McBride