Can We See Bonds with
X-Rays?
[Information in italics is not crucial to a qualitative
understanding of what's going on. It is just added in case you start
thinking too deeply for the simple explanations.]
Table of Contents (clickable)
1) Why can't one use a powerful light
microscope to see bonds?
2) Just HOW is structural information
contained in diffracted X-rays?
a) A single pair of scattering
points
b) An infinite row of evenly spaced scattering
points
c) A row of evenly spaced pairs of scattering
points
d) An hexagon of points ("benzene")
e) A pair of hexagons
f) A quartet of hexagons
g) A lattice of hexagons
h) Significance
3) What does Rosalind Franklin's x-ray
photo show about B-DNA?
a) Start by understanding the lightbulb
filament.
b) Base-Pair Stacking
c) Diameter
d) Double Helix - Major and Minor Grooves
4) What does the electron density in a
molecule look like? Are there Lewis Shared Pairs?
a) How to plot 3D electron
density
b) What do e-density maps say about molecules,
atoms, and bonds?
c) Accurate Difference Density maps show
bonds
5) Conclusions
1) Why
can't one use a powerful light microscope to see
bonds?
Light is ELECTROMAGNETIC RADIATION.
It consists of electric and magnetic fields that are oscillating
perpendicular to the direction in which the light is travelling (and
perpendicular to one another). For our present purposes we are
interested in the electric field, since that is the main actor in
pushing electrons around.
One can graph the oscillating electric field of light as a
sine wave, where the vertical axis is
the FIELD STRENGTH (+ or - means
pointing one direction or the other, i.e. exerting a force on
an electron in one directon or the other), and the horizontal axis
can be either TIME
or POSITION. That is, we can plot
the change of the field with time at any given position (perhaps
where a particular atom is), or we can plot the change of field with
position along the direction of light propagation at some given
time.
|
Graph of the force on an electron at some particular
position as a function of time due to the electric field
of a particular light wave. Of course the force on a proton
would be just the opposite.
One over the distance between successive maxima is the
frequency of the light.
|
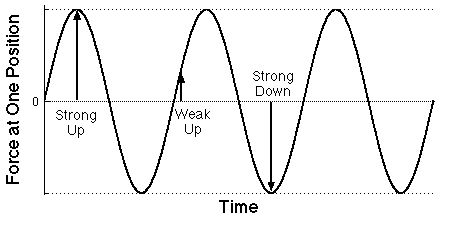
|
|
Graph of the force on electrons at some particular
time as a function of the position of each electron
measured along the direction that the light wave is moving.
Note that only the labels have been changed from the plot
above.
The distance between successive maxima is the
wavelength of the light.
As for a water wave, there are two different ways of
describing it with a wave-shaped plot.
|
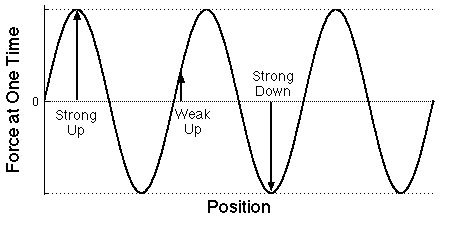
|
|
Graph of the force on the same electrons at a later
time as a function of the position of each electron
position measured along the direction that the light wave is
moving. The forces can change in both size and direction. As
time goes by, the wave moves to the right.
|
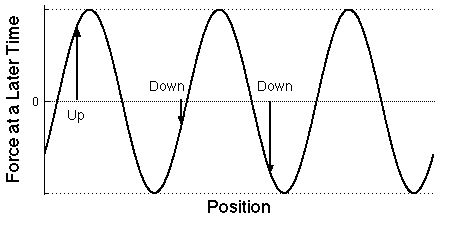
|
The electric field of light pushes charged particles of matter,
and the accelerating charges create new light waves that move off in
all directions, not just the direction in which the original light
wave is moving. This is called
SCATTERING. Of course the electrons,
being more than 1000 times lighter than the lightest nucleus, are
accelerated much more than the nuclei, so light
is scattered predominantly by electrons.
Thus if a light beam hits stuff and gets scattered, your eye can
detect light even though it is not in position to be struck by the
incoming beam. Deflected light is the signal that there must have
been electrons in the light path to do the scattering.
It is good to have a way of knowing that there are
electrons in the light path, but we want to know more. We want to
know where they are, how they are distributed, that is we want
to know the STRUCTURE
of the matter that is doing the
scattering. In particular, we want to know if electrons are
gathered around nuclei to form atoms and whether pairs of electrons
concentrate between certain pairs of nuclei to constitute Lewis
shared-pair bonds. This is related to the problem of
RESOLUTION (how far apart two things
have to be in order to tell that they are not a single piece).
One way to determine structure is to scan a VERY narrow beam back
and forth, up and down across the sample and see where it gets
scattered. This "raster "scanning (as in TV, or SPM) is used in
scanning electron microscopy and in an
SPM technique called SNOM (Scanning Near-field Optical Microscopy);
but beams can't be made narrow enough to resolve adjacent small
molecules, let alone atoms. Is there an alternative to scanning for
coding structural information into the scattered light? There must
be, because our eyes don't work this way.
|
Structural information is coded into an
INTERFERENCE PATTERN. Imagine
two electrons side-by-side in the path of the same incoming
wave so they are made to vibrate up and down in phase with
one another. Consider the oscillating electric field that
hits a detector at a very great distance [so distant
that there is no significant difference between the two
electrons in the angle by which the beam is deflected to hit
a certain spot on the detector]. The total field is the
sum of the two scattered fields, and because the path for
one is longer than for the other, depending on the angle,
the two waves will have different phase at any given time.
If the path difference is exactly one (or two, or three...)
waves, the fields will reinforce one another to give a field
twice as strong as that from one electron. If the difference
is half a wave, they will cancel. Thus as the deflection
angle increases from zero the amplitude detected for the
scattered wave will decrease, then increase, then decrease,
etc. How rapidly the amplitude of the
composite scattered beam changes with increasing
angle depends on the distance between the
electrons and the wavelength of the light.
Decoding this information about distance is the job
of the lens of the microscope (or the lens and retina of
your eye). We need not be concerned with the decoding, just
with whether the structural information is present to be
decoded.
|
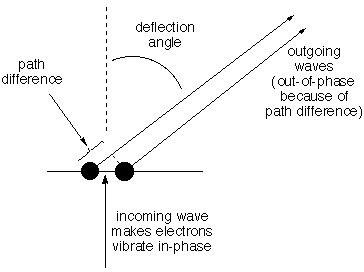
|
[The "wave machine" shown in class illustrated how such
interference arises. To be
picky I should note that we're ignoring
certain pathological situations where electrons in
different kinds of atoms may not vibrate precisely in phase with one
another even when they are being struck at the same time by the same
wave. This situation is called anomalous
dispersion, and can be quite useful.]
|
If the wavelength is much greater than the distance
between the electrons, there can be no significant
difference in phase for any scattering angle, and
thus no clue in the scattered light that there are two
separate electrons. We can only
resolve objects whose separation is comparable to, or larger
than, the light wavelength.
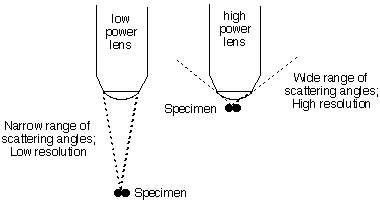
[Note that the greatest phase difference occurs for
light scattered at 90° from the incoming ray. Thus to
get maximum resolution from light of a given wavelength one
must collect scattered light up to a wide angle. This is why
the objective lens of a high-power microscope must get very
close to the object in view.]
|
|
The shortest visible wavelength is >400 nm = 4000 Ĺ ~ 2700
times the C-C bond distance. So forget using light microscopes to
resolve adjacent atoms within molecules or even adjacent molecules of
reasonable size.
If we want to resolve atoms, we must use waves that are about 1.5
Ĺ (0.15 nm) long. This means
X-rays, but there is no good lens for
focussing x-rays, so we must use a computer to decode the structural
information from the interference pattern collected over a wide range
of angles.
[High-kinetic-energy electrons also behave as waves
with short wavelength, and there are electric "lenses" for
them. They can be used for transmission electron microscopy
(TEM), but they are much less
generally useful than x-rays, because electrons are so strongly
scattered that they can't go through air or through more than a
few molecular layers of a sample.]
2) Just
HOW is structural information contained in diffracted
X-rays?
It is easy to appreciate that, if the wavelength of the light is
short enough (x-rays), scattered light contains information about how
electrons are distributed in a sample. [A good analogy is a water
wave which encounters the pilings in a pier and generates a complex
pattern of reflected waves that depends on how the pilings were
arranged.]
The problem is how to decode the information.
Nowadays sophisticated computer programs can do the decoding
pretty easily for crystals of even rather large molecules - most
recently and dramatically (and at Yale) the ribosome with hundreds of
thousands of atoms. For most people these programs are just black
boxes. It is rewarding to think a little about how the information is
contained in the scattered X-rays. This is why we used the He/Ne
laser (wavelength 632.8 nm) in the lecture demonstration.
[First we think of scattering within a single plane, as
visualized with the "wave machine". The first slides used in the
lecture demonstration consisted of vertical lines which, viewed from
above look like the "points" discussed below. I give this technical
information for the sake of honesty - you may stick with 2 dimensions
and think of points instead of lines to get the idea. It is cute that
in 3-D, just as a set of parallel lines on the slide give a row of
dots on the screen, a row of dots on the slide gives a set of
parallel lines on the screen.]
|
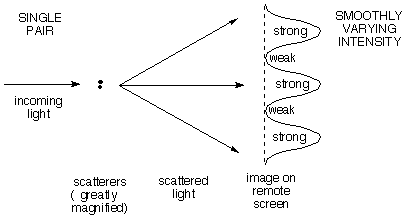
a) A
single pair of
scattering points (e.g. electrons) gives an intensity
distribution that varies
smoothly with
increasing scattering angle from maximum (straight
ahead) to minimum to maximum.
The angular distance between maxima (together with the
wavelength) tells how far apart the points are. Note the
RECIPROCAL
relationship: the closer the points are together, the larger
the angular difference between scattered maxima. The
first deflected maximum comes when the wavelets from the two
scatterers differ by one wavelength, the second when they
differ by two, and so on.
|
|
|
You are probably familiar with this case already as the
"two-slit" experiment in which a plane wave of light
travelling left to right passes through two slits viewed
end-on 1/4 and 3/4 up the left side of the diagram. The
resulting waves interfere (add to one another) to give at
some instant in time the pattern shown. The light areas are
peaks and the dark areas are troughs. Note that as the light
moves far to the right the patterns coalesce into a set of
rays fanning out from a point between the slits, and
separated by gray areas of zero electric field. This gives
the smoothly varying intensity shown on the remote screen in
the diagram above. If you'd like to see an
animation of these interfering
waves, click for the applet at the Physics
2000 website.
|

|
|
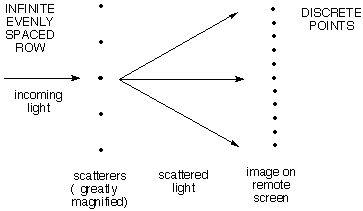
b) An
infinite row of
evenly spaced scattering points gives a row of
discrete
scattered spots.
If the angle is chosen so that the waves scattered from
the first two points differ by exactly one wavelength, the
wave from the third will differ from that from the first by
two wavelengths, etc. So all the waves will be effectively
in phase to give very strong constructive interference. If
the angle is changed just a little bit so that the first two
waves are not exactly one wavelength different, the next
will differ by twice as much, and so on, so that ultimately
cancellation will set in.
|
|
For example, if waves from the the first two scatterers differed in
phase by only 1% of a wavelength, the 50th would differ from the
first by half a wavelength, and they would cancel. The 51st would
cancel the 2nd, and so on. So there is a lot of blank space between
successive maxima. As above, the angular distance between maxima
(together with the wavelength) tells how far apart adjacent points
are, and the relationship is reciprocal, large spacing of
scatterers means close spacing of spots in the image.
The problem assigned in class was to determine the
spacing between the rows on the slide that would give diffraction
maxima separated by 10.8 cm on a screen at a distance of 10.6 m
when the wavelength is 633 nm. (the answer is about 65 microns).
Doing the geometry will confirm your understanding of the source
of interference. Consider whether the points in the image should
be spaced exactly evenly.
|
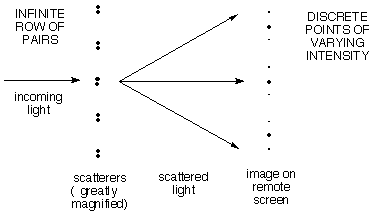
c) More interesting
is a row of evenly spaced PAIRS of scattering points,
where the pairs are closer together than the repeat distance
between pairs.
Here the pattern is the row of closely-spaced dots
expected from (b) for the large repeat distance
between pairs (remember the reciprocal relationship between
spacing and angle), but the intensities vary slowly as
expected from (a) for the smooth variation due to the
members of a single closely-spaced pair.
|
|
Here's how to think about this:
The pattern which is to be repeated generates
a smooth variation of scattered intensity that tells about the
structure of the pattern.
Here the pattern is a pair of dots. We know above
that any pair of dots with this spacing will scatter with a
smooth variation in intensity as shown in (a) above. Since
all of the pairs have the same spacing, they will all
scatter with the same angular dependence. Now all we have to
worry about is how the scattering from one pair interferes
with the others.
The fact of the regular repetition allows one to observe
this scattered intensity only at the discrete positions allowed
by the repeat distance. [Importantly, the net scattered
intensity is unchanged. It is as if the intensity of the
diffuse pattern is gathered into the spots. This makes the
spots easy to measure.]
This is like viewing the smooth distribution due to the pattern
through little holes whose spacing is determined by (and
reciprocal to) the row repeat, but the intensity is amplified.
|
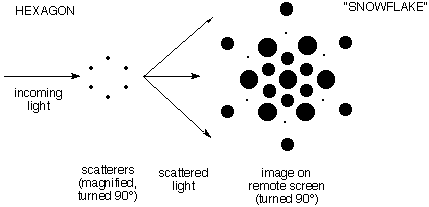
d) The second
demonstration showed scattering in three dimensions. It
involved a hexagon of points ("benzene"). A single
set of six points generated a smoothly varying "snowflake"
pattern of scattered intensity. The following illustration
gives an idea of the diffraction pattern, but fails to shows
how smoothly it varies.
(Note: In this schematic illustration the
pictures of the scatters and the image are turned
90° so you can see them. Actually they lie on planes
perpendicular to the ray of incoming light.)
Click
here to see the Laser Demonstration
|
|
[In order to get enough intensity to be visible the slide
actually had an enormous number of hexagons, all oriented the same
way but randomly placed. Randomly placed patterns give the same
diffraction as single patterns, but more intense. This contrasts with
a lattice of regularly placed patterns, which focusses the scattering
into discrete points, as described in e-g.]
|
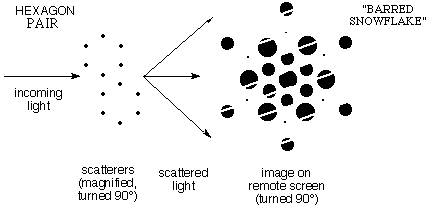
e) When the
pattern contained pairs of "benzenes" the diffraction showed
the same snowflake pattern but with intensity varying along
the direction in which the pairs are displaced.
Click
here to see the Laser Demonstration
|
|
|
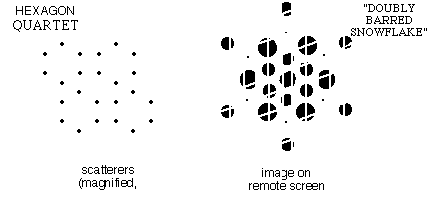
f) When the
pattern contained quartets of "benzenes" at the corners of a
parallelogram, the same snowflake pattern was modulated in
intensity in two directions.
Click
here to see the Laser Demonstration
|
|
g) When all of the
"benzenes" in the slide fell on a regular lattice with
standard row and column spacing, the diffraction pattern showed the
intensity distribution of the original snowflake but viewed only on
the two dimensional lattice of points allowed by the regular spacings
in rows and columns. This is the 2-dimensional analogue of (c)
above. It is as if the snowflake were being viewed through the
holes in a piece of pegboard.
Click
here to see the Laser Demonstration
h)
Significance:
The LOCAL ELECTRON DISTRIBUTION
(molecular structure) is coded in the
INTENSITY of the spots (in this case,
the underlying continuous snowflake pattern).
The SPACING OF THE ROWS AND COLUMNS
(crystal lattice) is coded in the
ARRANGEMENT of the spots (the "pegboard"
holes through which the snowflake is seen).
X-ray diffraction is usually measured with
single crystals. The crystal serves
two purposes:
First, it orients all of the molecules in the same
way, so the snowflakes reinforce.
Second, it concentrates all of the scattered light into a
small number of points which are bright enough to observe
easily.
3) What
does Rosalind Franklin's x-ray photo show about B-DNA?
|
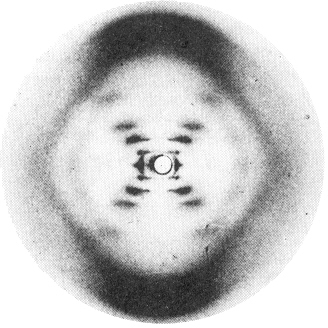
The diffraction of He/Ne red light by a slightly
stretched filament from a broken lightbulb shows an "X" with
spots along each leg (see below).
This dotted "X" bears a strong resemblance to Franklin's
photo (right) of DNA fibers in which molecules were oriented
more or less vertically by stretching.
Since neither the filament nor the DNA is crystalline,
both show the rather diffuse pattern from an individual
object (like a pair - or hexagon - of dots) rather than the
set of sharp spots characteristic of an infinite regular
lattice of such objects.
Of course there is a certain degree of repetition within
the helix itself, which gives rise to the features that are
discussed below. These helices are great examples of how
molecular structural information is present in the
scattering pattern. For example, the regular repetition from
turn to turn along the helix axis constitutes a repeating
vertical pattern that gives rise to horizontal bars, as in
2c or 2e above.
|
|
a)
Start by understanding the lightbulb filament.
|
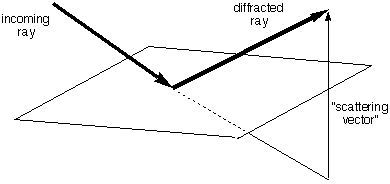
We need the following rule:
All electrons on the
same
plane
perpendicular to the
scattering vector
(the change in light direction, see figure) scatter
with the
same
phase.
[This is in fact how a mirror works - all
the silver atoms scatter light in phase when the incoming
and outgoing beam angles are the same.]
For purposes of calculating the scattered intensity, all
the electron density in such a plane could all be considered
as concentrated at any single point on the plane
perpendicular to the scattering vector. THIS IS A VERY NEAT
TRICK.
[It is easy to prove this rule
geometrically by showing that all the path lengths (sum
of incoming and outgoing) are the same]
|
|
|
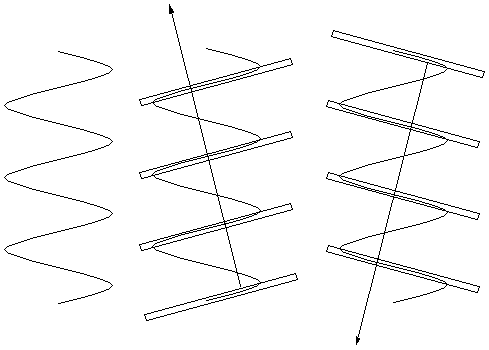
So we can look for planes in the sample that have a lot
of electron density and think about the light that is
scattered (or "reflected") from them.
For a vertical helix (right) there is a lot of electron
density on the slanted planes shown (middle and far right),
and not much on any given parallel plane between them.
Thus the indicated scattering vectors should be imporant.
That is, some of the light going perpendicularly into the
page should be deflected up to the left (and down to the
right) by the "planes" highlighted in the center figure.
Some should also be deflected down to the left (and up to
the right) by the "planes" highlighted in the right figure.
These are the directions of the arms of the "X".
|
|
[Actually the helix must be slightly tilted to
bring these particular planes into reflection position. This may
be taken care of by rocking the sample a bit during exposure of
the film, or, as in the Franklin case, by the fact that different
molecules in the fiber have slightly different orientations so
that many have the desired orientation of slight tilt in any given
direction. Even without the axis tilting, there will be an
analogous set of planes slightly further along the helix (and a
few degrees around to the right) that will be tilted into the
correct position. This is a fine point - don't worry about it if
you don't see it easily. There are more important things to think
about.]
For scattering in the indicated directions the e-density on the
planes could be thought of as concentrated at the points where the
arrow intersects the planes. That is, there is scattering as if from
a row of dots that would reinforce at a given succession of angles
(as in 2b above). There is very little electron density on the
interleaving planes where it would have to be to cancel these
reinforcing waves.
Thus the "X" of dots in Franklin's photo shows that DNA
is a helix, and the angle of the "X" shows how tightly it
is wound (think about this).
Click
here to see the Laser Demonstration
b)
Base-Pair Stacking
Successive units of the DNA polymer are spaced at a given distance
along the axis of the helix. For example, one can see planes that
contain practically all of the carbon and nitrogen atoms of the
"bases" in stacked planes perpendicular to the axis. These planes are
much closer together than the "helix planes" illustrated above
so they should reinforce for scattering at a much higher
angle. This gives rise to the very dark spots at the top and
bottom of the Franklin picture and shows that the stacking distance
is 3.4 Ĺ (the thickness of an aromatic ring).
|
c)
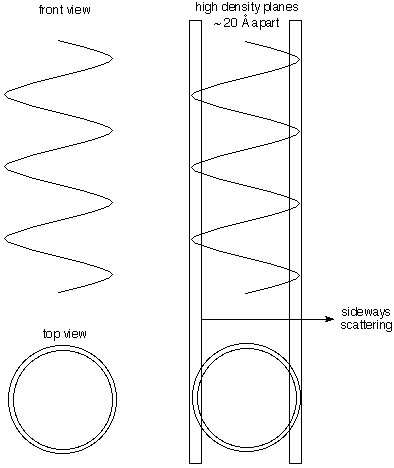
Diameter
Much of the electron density of DNA is in the phosphorous
and oxygen atoms on the helical chain. Thus lots of the
electron density lies on the periphery of a cylinder. The
illustration to the right shows that planes that are nearly
tangent to the cylinder contain lots more electron density
than those halfway between them (because the center is not
so dense in electrons. This gives lateral scattering that
measures the 20 Ĺ diameter of the helix (the spots
look like triangles in Franklin's photo).
|
|
d)
Double Helix - Major and Minor Grooves
Other things being equal, spots get less intense as they move to
higher scattering angle. This is observed as one moves out along the
arms of the "X" from the lightbulb filament scattering.
Things are different for DNA. As one moves out from the center the
1st spot is rather weak, the 2nd and 3rd are strong, the 4th is too
weak to see, and the 5th is strong enough to see clearly - much
stronger than the fourth. What a queer sequence of
intensities!
[Incidentally, the undeflected beam would be so
strong as to wash out everything else - therefore a lead cup is
put in its path to intercept it, so there is no spot in the center
of the photograph.]
|
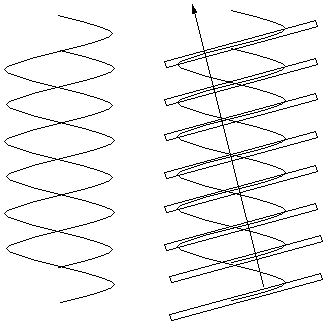
The intensities show that DNA is a double helix
with a major and a minor groove
Had the structure involved an evenly spaced double
helix (as shown right) the spots on the "X" would be
twice as far apart (i.e. every other spot from the single
helix would be missing), because the slanted planes of
electron density would be twice as close together.
Another way of saying the same thing is the
following:
For a scattering angle where waves from successive
planes in the same helix differ by an odd number of
cycles (and thus reinforce one another), those from the
other helix differ from them by a half-integral number of
cycles and cancel the waves from the first helix.
When reflections from successive planes of a single
helix differ by an even number of light cycles,
reflections from planes of the other helix differ from
them by an integral number of cycles and reinforce.
|
|
|
But what if the helices are offset from even spacing to
give major and minor grooves as shown to the right?
The pattern of planes is like that of the spots in
example 2c above - a widely spaced row of
closely spaced pairs of scatterers. The resulting
diffraction pattern would also be the same - a row of points
spaced as for the row (single helix) spacing, but with
modulated intensities (weak, strong, strong, very weak,
intermediate) that can be used to measure the amount of
offset between the two helices. This is exactly what the
Franklin photograph shows.
|
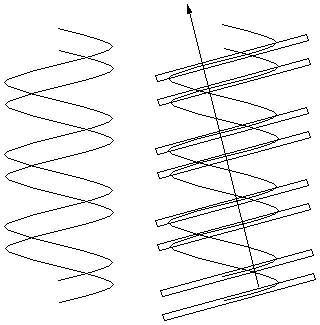
|
4) What
does the electron density in a molecule look like? Are there Lewis
Pairs?
a) How do you visualize three dimensional plots
of electron density?
Electron density is a function of three variables - x, y, and z
positions. How can you plot it?
On a 2-dimensional sheet of paper it is convenient to plot two
things against one another (two variables - independent and
dependent). It is harder to plot a function of two variables,e.g.
altitude (or temperature) as a function of latitude and longitude.
With color or a gray scale it is possible to plot such a function of
two variables. Another convention is to use a contour plot where
lines connect two-dimensional locations of a given value. You should
be familiar with such plots of isotherms (regions of the same
temperature) and with topographic maps showing a mountain as a series
of nested curves.(click
here for a brief exercise on 3D plots)
Plotting a function of three variables is much more difficult. One
way would be to take a big block of clear jello and use an hypodermic
syringe with dye to inject more or less color in regions of certain
electron density. One could generate an image of such an object using
3-D computer graphics (this is precisely what Dean Dauger did to
represent the electron density in hydrogen-like atomic orbitals in
his program Atom in a Box, which we'll use soon). A practical
scheme for presentation on paper is to make a well-chosen cut through
3-D space to define a particular plane of interest (for example a
plane containing the atomic nuclei of a planar molecule) and then use
a contour plot to show electron density on that single plane. One
could make a lot a parallel planar cuts and show a bunch of such
contour graphs. No one said it would be easy. (here
is an example of such a planar map)
|
The figure to the right shows contours on part of an
arbitrary slice through an e-density "map" from an X-ray
study of Rubofusarin.
Numbers giving the e-density were typed on a page at the
appropriate x,y positions by a computer and the
crystallographer then connected locations of the same value
with a pen to generate contours.
This was about 40 years ago - before it was easy
and cheap to make computers draw solid curves. There were
no laser or ink jet printers or graphic (or color)
terminals. Electric typewriters or close relatives were
the common output devices. Imagine!
From examining the numbers it looks to me as though they
were contouring at values of 150, 200, 250, 300, 350, 400
(in arbitrary units, not e/Ĺ3).
(from Stout and Jensen "X-ray
Structure Determination" 1968)
|

|
|
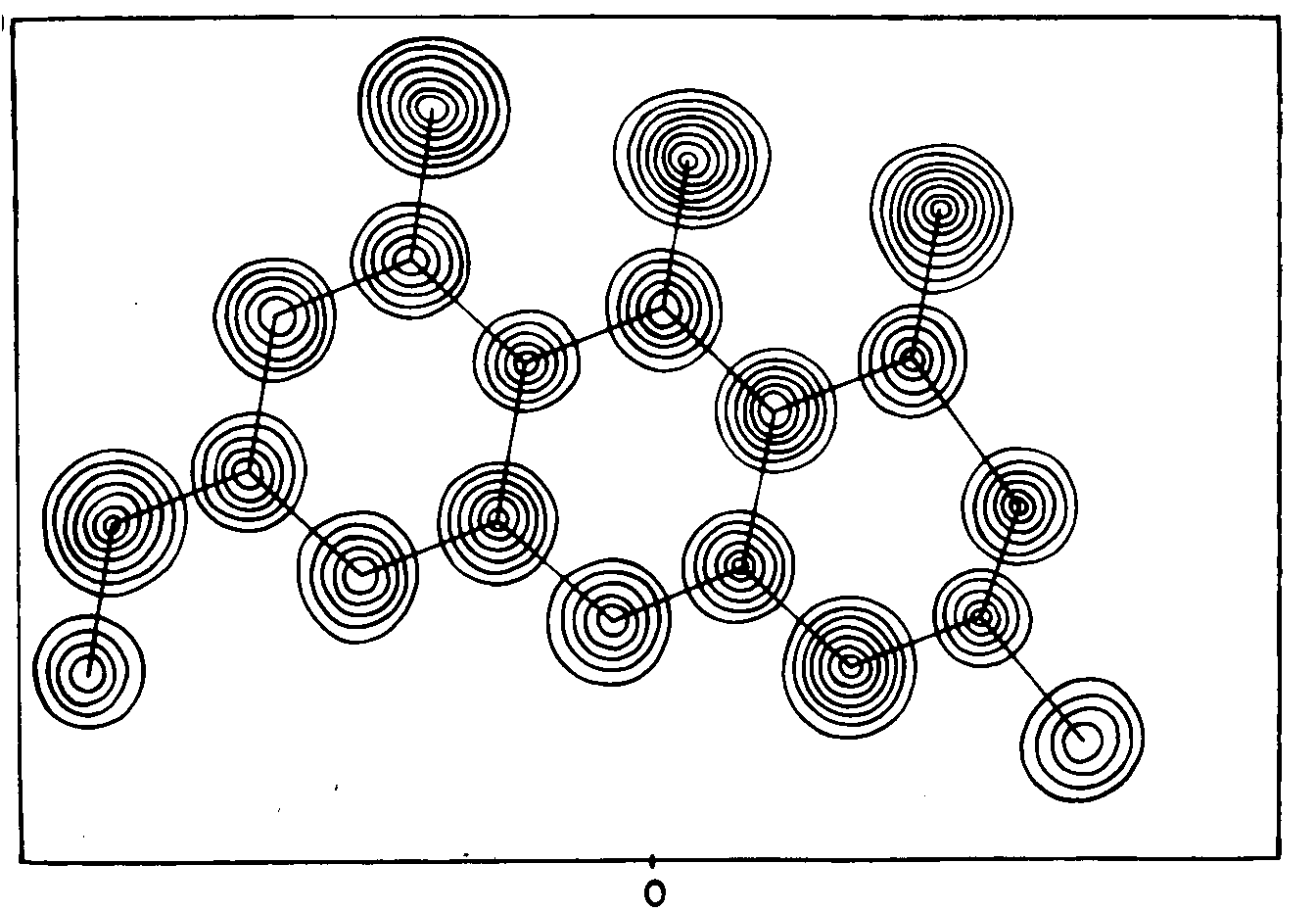
Not all of the atoms fell on the slice plotted above.
Fortunately the molecule of Rubofusarin is nearly planar,
so the crystallographer could calculate an analogous sheet
of numbers for a slice chosen to contain most of the
molecule's atoms.
He then traced a similar set of contours on an overlaid
sheet of plain paper to give the figure shown to the right.
Here he drew contours at intervals of 1
e/Ĺ3.
|
|
|
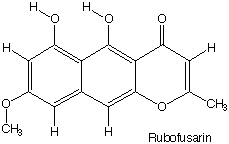
Correspondence of the spheres of high electron density
with the non-hydrogen atoms in the formula of Rubofusarin is
obvious.
|
|
Rubofusarin has the advantage of being nearly planar, so that a
single planar slice can give a good idea of the electron density of
almost all of its atoms. In a non-planar molecule one must choose
different slices to show features of the electron density with a
two-dimensional contour map. One approach, used in the 1940s in
connection with determination of the structure of the potassium salt
of penicillin by Dorothy
Crowfoot (later Hodgkin)
at Oxford, is to draw contour maps for a set of sequential, parallel
slices on transparent sheets and stack them up to give a
three-dimensional contour map, actually a sort of four-dimensional
graph! [KPenicillin
illustration]
b) What do e-density maps say about molecules,
atoms, and bonds?
There is a lot of information in this e-density map.
Perhaps the most important is that this organic
molecule looks like a collection of
spherical atoms with electron density increasing toward
the nucleus of each. There is certainly no "double dot" of
electron density between the atoms. In fact the spheres don't even
look distorted. There is no evidence from this plot to support
Lewis's idea that bonds consist of shared electron pairs!
Note that electron densities identify the atoms:
hydrogens have too little electron density to be
visible in this map (less that 1 e/Ĺ3)
only oxygen atoms achieve electron density greater than 7
e/Ĺ3 (seven contour lines).
The nuclei don't appear directly in the map, since they
are too heavy to scatter x-rays significantly, but one can guess
that they are located where the e-density is highest at the center
of each sphere. [This inference can be confirmed by
experiments involving neutron scattering - neutrons are scattered
by nuclei and thus can reveal their position
directly.]
The bond lengths show what kind of bond is involved:
|
Carbon-Carbon
|
Dist (Ĺ)
|
|
Carbon-Oxygen
|
Dist (Ĺ)
|
|
Single
C-C
|
1.55
|
|
Single
C-O
|
1.37
|
|
Double
C=C
|
1.32
|
|
Double
C=O
|
1.25
|
Such e-density maps confirm the reality of molecules and atoms but
not of the Lewis shared-pair bonds.
c) Accurate Difference Density maps show that
electrons DO shift (a little) to make bonds.
Of course one can wonder whether the apparently spherical electron
density distributions that look so much like isolated atoms might be
at least a little bit distorted. The most sensitive way to check this
is with a "charge-density difference map". Instead of plotting the
total electron density, one plots the difference in density between
what is observed in very accurate experiments and what would be
expected for an analogous collection of perfectly spherical,
undistorted atoms. If there is no distortion upon forming bonds, the
difference density should be zero everywhere, but if electron density
shifts a little, there will be positive regions where density
accumulates and negative regions where it is depleted.
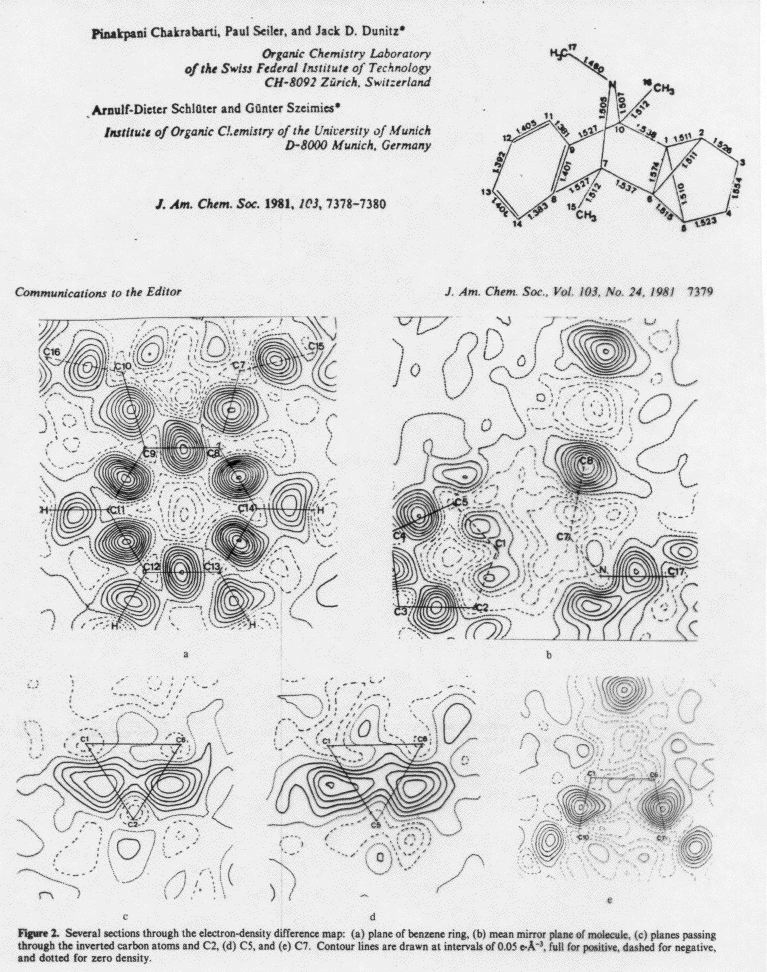
One lecture overhead showed such a difference density map for a
molecule with single C-C bonds, aromatic C-C bonds, CN triple bonds,
and C-F single bonds. Solid contours show where electron density
increased upon forming bonds, and dashed (negative) contours show
where it was depleted. Notice that the amount of density shifted is
very modest - the contour level in the difference map is only 0.075
e/Ĺ3, less than one tenth of the 1
e/Ĺ3 interval used in contouring the total e-density
map above. Even in the triple bond the maximum increase in e-density
between the nuclei is only 0.7 e/Ĺ3, less that 10%
of the density near an oxygen nucleus. [Tetrafluorodinitrile
overhead; see Lecture 6]
Lewis might have been gratified to see that upon forming the
molecule e-density accumulates between the atoms in "bonds", and in
an unshared pair on the nitrogen atom. [He had died 20 years
before experiments of sufficient accuracy were possible.] It
is also satisfying to see that the greatest accumulation of e-density
is in the triple bond, followed by the "resonant" aromatic
"one-and-a-half" bonds, and the single bonds. [The much more
modest accumulation in the C-F bonds is surprising and requires some
mental gymnastics to rationalize. (Interview
with J. D. Dunitz)]
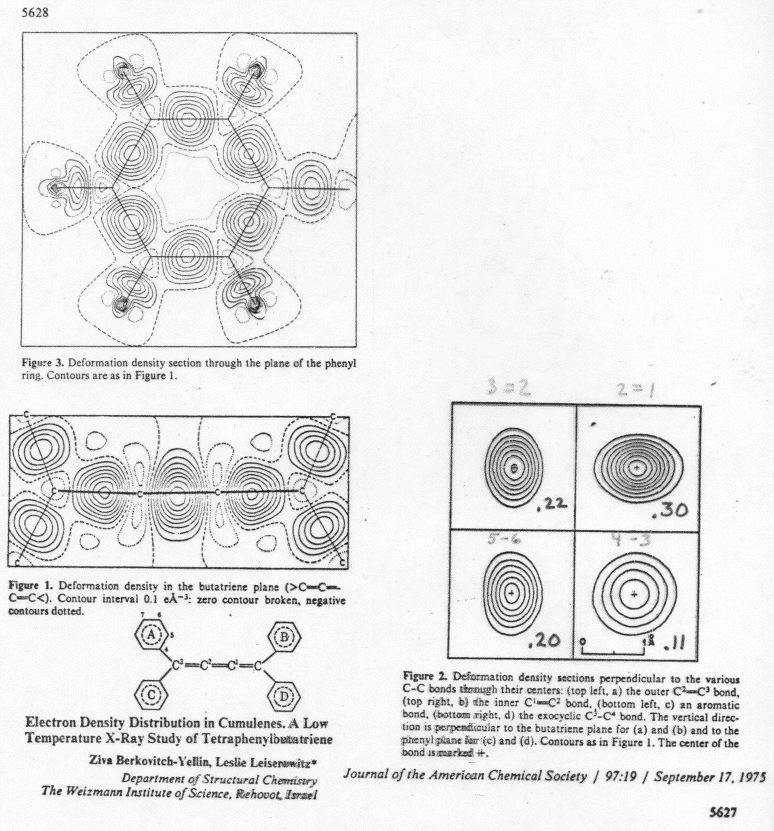
Perhaps even more striking is the cross sections of the
bonds. The single and triple bonds have circular cross sections,
while the aromatic bonds have oval cross sections consistent with the
contribution from both sigma and pi bonds (which we will discuss a
lot more later). [Cumulene
overhead, see Lecture 6; Interview with L.
Leiserowitz] [Cyclopropane
overhead, see Lecture 7]
5)
Conclusions
So, using x-ray vision, we can see
experimentally that molecules
and atoms are real, and that bonding is associated with (modest)
accumulation of electrons between the bonded atoms.
Here is the answer to "How do you know?" - not because some
teacher or book told us there are atoms and bonds, but because it
seems plausible that previous scientists accurately reported x-ray
experiments of which we understand the significance. If we were
skeptical, we could repeat the experiments and see for ourselves.
Our next task is to understand WHY
the electrons gather into bonds and what properties bonds should have
(length, angles, strength, reactivity), and more fundamentally what
is special about octets, why there are electron shells, etc.
On
to Quantum Mechanics!
[Return
to Chem 125 Home Page]
latest revision 9/10/05
Comments on this page are welcomed by the author.
J. Michael McBride
Department of
Chemistry, Yale University
Box 208107, New Haven, CT 06520-8107
e-mail: j.mcbride@yale.edu
copyright
© 2001,2004,2005 J.M.McBride
{kind=link}
{kind=link}
{kind=link}