|
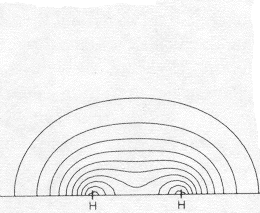
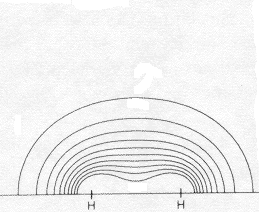
contour at 0.025 e/ao3 |
Bond Density Bond Energy |
contour at 0.005 e/ao3 |
 |
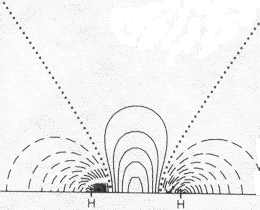
1s (atomic) electron repulsion adjustment: Density at bond center: Difference at bond center: Fraction of true Bond Energy |
 |
|
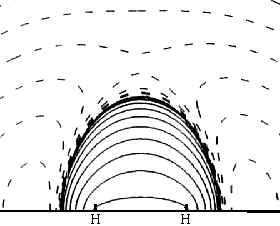
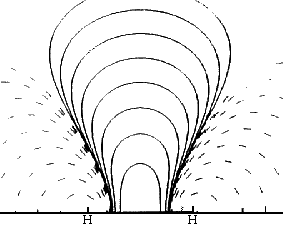
This very simplest guess for the MO of wave functions for the two bonding electrons is <2-1/2 (1sA + 1sB) , where 1sA and 1sB are the true 1-electron wave functions of isolated H atoms (centered on the two nuclei and taken from the Table we have used). It does a pretty good qualitative job of showing an accumulation of e-density in the bond and a lowering of energy from that of two isolated atoms. In the difference map note how electron density shifts from the atoms (dashed negative contours) to the bonding region (solid positive contours). Quantitatively it's not so good. It shows only about 10% of the true increase in bonding difference density, and only about half of the bond energy. The bond energy is the smallish difference between the substantial stabilizing energy of the isolated atoms (vs. separated electrons and protons) and the energy of the molecule. Naturally the approximation does a better job on the total energy (93%) than on the small bonding energy, since most of the energy, like most of the electron density is just that of the two separate atoms. |
||
|
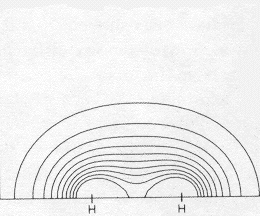
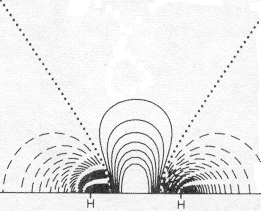
expanded 1s electron repulsion adjustment: Density at bond center: Difference at bond center: Fraction of true Bond Energy:
|
|
|
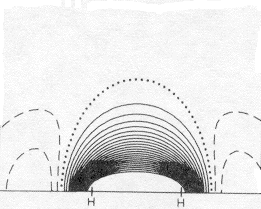
Use the same 2-1/2 (1sA + 1sB) expression above, but employ the simple trick of adjusting the constant multiplier, K, of the exponent in the 1s atomic orbitals (C e-Kr) until the energy of the MO is minimum. Allowing the atomic orbitals to spread out a bit in this way nearly doubles the bonding electron difference density and halves the error in the bond energy from 48% to 27%. But simply spreading the 1s function increases bonding density at the expense of electron density near the nuclei. |
||
|
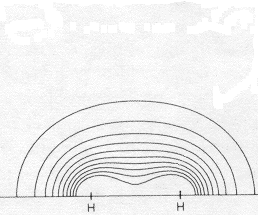
96.7% 1s; 0.6% 2s; 2.7% 2p electron repulsion adjustment: Density at bond center: Difference at bond center: Fraction of true Bond Energy:
|
|
|
Here the energy has been minimized by hybridizing a bit of 2s and 2p into the 1s descriptions (again with exponents optimized, this time taking e-repulsion into account in optimizing the orbitals via SCF). Of course one cannot hybridize much without spoiling the description of the electron distribution in the vicinity of the nuclei, since for the atom the 2s and 2p orbitals are much higher in energy than the 1s orbital. [Note that the "atomic penalty" for hybridizing 2p with 2s orbitals in carbon will be much lower than for 2p with 1s in hydrogen, since orbitals with the same principal quantum number have very similar energies.] It is remarkable how drastically only 3% of a higher orbital can change the description, increasing bonding difference density by almost a factor of three, although the energy doesn't change much. It is not surprising that the best hybridization has much more 2p than 2s character. Consider why 2p is particularly helpful in making the atomic orbitals spread out in the right direction, rather than just moving electron density away from the nucleus in all directions as in previous case. |
||
|
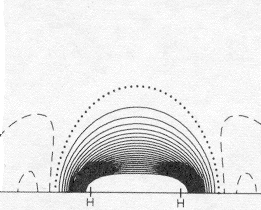
1s ; 2s ; 2p electron repulsion adjustment: Density at bond center: Difference at bond center: Fraction of total Bond Energy:
|
|
|
Taking some account of electron correlation (allowing the two of them to stay apart) yields a big improvement in the energy without changing the overall electron density much at all. The "correlated" wave function is written so that when one electron is in a region of high density, the other one can be in a different region of high density. The bond strength is now within 10% of correct. Because of computer software and hardware advances over the intervening 25 years one can now do a better job, with modest effort, on much more complex systems, including most simple organic molecules. For quantitative purposes one increasingly depends on such numerically intensive calculations, but for qualitative purposes we can see that the very simplest <2-1/2(A+B) function doesn't do too bad a job of showing why there is a bond in H2, and how hybridization helps. |
||
|
|
with a different set of contours Left: Difference density with Right: Difference density after Thanks to Prof. Paul Rablen |
 |
|
|
|