Visualizing Molecular Orbitals with
MacSpartan
In trying to understand molecular orbitals you
will almost certainly be helped by viewing them in color and in
motion using computer graphics (especially if you find the pictures
of MOs that we show in class or on handouts confusing). If you happen
to have blue/red stereoglasses, you can even see them in
3D.
There are a number of programs that allow you to
do this with orbitals, but one that is readily available on the Yale
Mac clusters is MacSpartan PLUS. There is a PC version of this
program, but we don't have it. Sorry.
With MacSpartan PLUS you can construct molecules
and do MO calculations on them at various levels of sophistication up
to SCF with reasonably flexible atomic orbital shapes
(6-31G(*)). At present we will only be using it to
view orbitals that have already been calculated. They are available
in the Chem125 MOs folder in the MacSpartan Plus
folder.
You'll find the program at the following location:
MacHardDisk / Instructional Software / Science/Math
Here's what you need to do once you have launched
MacSpartan PLUS by double clicking its icon.
Choose an appropriate background color by
clicking the Apple at the top left and selecting Set Colors. Nothing
very wrong with white, but black may be more artistic.
Under FILE choose OPEN, click to get the
Chem125MOs folder, and choose one of the five subfolders (HF,
CH3F, C2H5F, H2C=O, CH3, CF3) within it. I suggest you start with
HF. Then click the file with the same name and you're in
business. A framework model of the molecule will appear on the
screen.
You can adjust the type of model with
options under the MODEL menu. Try the following:
Wire
Ball and Wire,
Tube,
Ball and Spoke, and
Space Filling
You can further adjust the view by clicking
and moving the mouse with the help of various keys.
Just click and drag to rotate in and out
of the plane
Hold clover while you click and drag up and down at the left of
the screen to rotate about the axis of view
Hold option clover while you click and drag up (or down) to
magnify (or compress) the view
Hold option click and drag to shift the molecule within the
frame
For best viewing the orbitals, increase the size
of the model and choose Ball and Wire or Tube under
MODEL.
Now go to the DISPLAY menu and click
SURFACES. Double click one of the orbitals in the box at the
top of the new window. A check mark appears at the left to show you
want to display this orbital, for example HOMO-1. Several can be
checked and shown simultaneously, but this gets confusing. Specify a
STYLE of display (Solid is the default, but I prefer Mesh or
Transparent). Click OK and you can look at the orbital in "color"
(what do the colored surfaces specify?) and space (perceived by
rotation with the mouse).
|
For most molecules only one surface has
been calculated, the most interesting one that is
characteristic of the functional group. To look at other
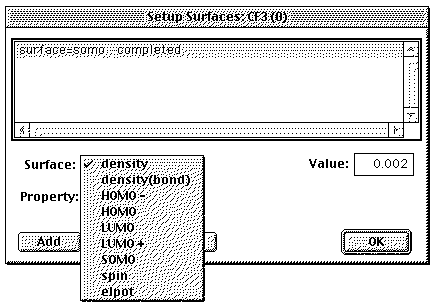
orbitals go to SETUP / SURFACES and select what you
want.
For example clicking Surface
density and leaving Property as none, as shown
below, would generate a contour where the total electron
density is 0.002 e/ao3, which
corresponds approximately with the van der Waals envelope
(the "size") of the molecule. Choosing density (bond)
makes the contour at 0.08 e/ao3. (for
density you can choose other contours with Value,
in fact you can display several contours simultaneously,
onion style, by having the largest value, i.e. the smallest
contour displayed solid, the intermediate one as a
mesh, and the outer one as transparent
).
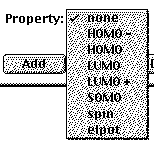
Normally Property will be
none, but choosing elpot adds surface coloration
coding the energy that an electron would have at different
points on the surface. This shows something about how charge
is distributed in the molecule.
What we're mostly interested in now is
the different orbitals, so usually you'll just name a
Surface like HOMO-2, and choose none for the
property. After you've made your choice click Add,
and it will appear in the list with the designation
{pending}. After you've chosen all the surfaces you want
to look at, click SETUP / SUBMIT and in a few 10s of seconds
with a fast Mac the surfaces will be calculated.
|
|
|
|
You can then examine your new surfaces with
DISPLAY / SURFACES, as before.
Have a good time, and remember to quit the
program when you're not using it actively.
return to
Chem125 Homepage